
Introduction à la MH et au CAG
La MH est une maladie neurodégénérative progressive autosomique dominante causée par une expansion de la séquence CAG dans le gène HTT situé sur le chromosome 4. Les individus en bonne santé ont entre 10 et 35 répétitions CAG, bien qu'il existe des preuves que ceux avec 27 à 35 répétitions CAG, connu comme allèles intermédiaires, peuvent montrer des signes d’apparition plus tard dans la vie (Groen et al. 2010). Les porteurs de l'expansion du gène MH (HDGEC) ont 36 répétitions CAG ou plus. Ceux avec 36 à 39 répétitions ont une pénétrance réduite, ce qui signifie que certains développeront des symptômes de MH au cours de leur vie, tandis que d'autres ne le feront pas (McNeil et al. 1997). Les personnes avec 40 répétitions CAG ou plus ont une pénétrance complète et développeront la MH au cours d'une vie normale.
L'âge auquel les HDGEC présentent l'apparition des signes moteurs est associé à la longueur de la répétition CAG, et ceux qui ont plus de 60 répétitions CAG sont susceptibles de présenter des signes très tôt et de développer une MH juvénile avant d'atteindre l'âge adulte (Fusilli et al. 2018). Dans les études observationnelles MH, la longueur de CAG la plus courante est de 42, la plupart des HDGEC allant de 40 à 44 CAG. La distribution des longueurs de CAG pour Enroll-HD est illustrée dans la figure 1.
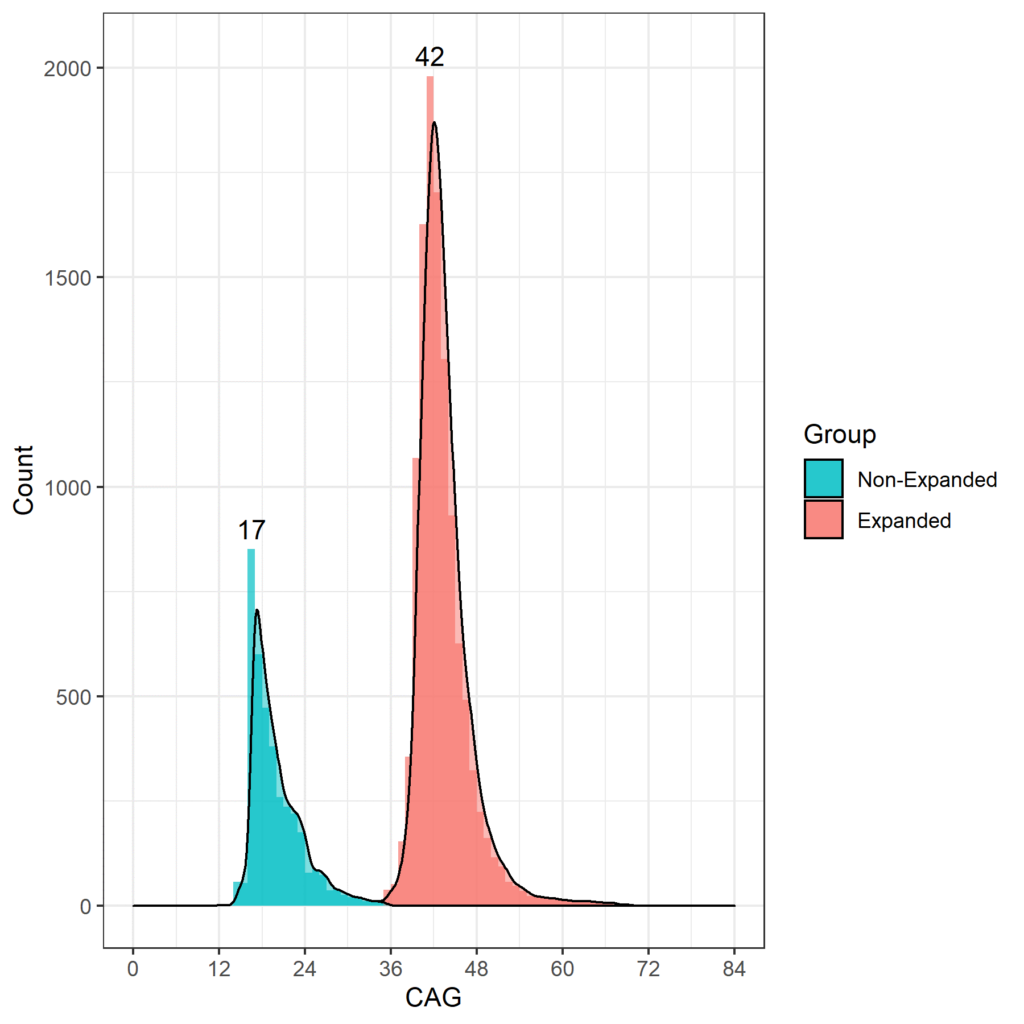
Figure 1. Répartition des longueurs de CAG dans la population Enroll-HD. Répartition des longueurs de répétitions CAG chez les non-HDGEC et les HDGEC dans Enroll-HD illustrées séparément. Valeurs modales pour chaque groupe explicitement répertoriées. (version PDS4 ; v2018-10-R3).
Apparition et diagnostic de la MH
L’apparition et le diagnostic de la MH sont des concepts importants d’un intérêt crucial pour de nombreux chercheurs. Dans cette section, nous discutons en détail de ces concepts complexes et des nuances des variables qui sont collectées dans l'étude Enroll-HD.
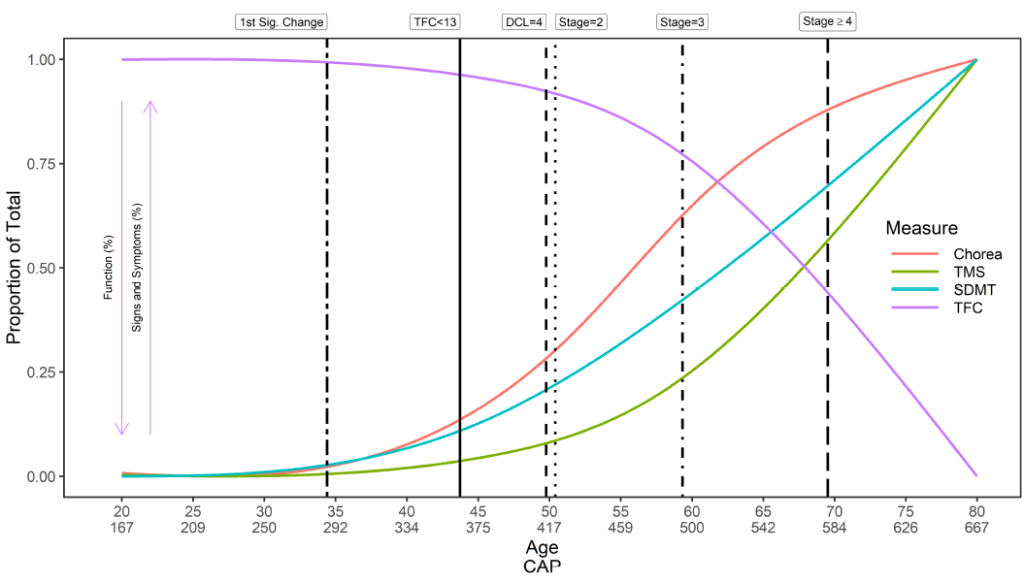
L’apparition de la MH est complexe. Un aperçu générique de l'histoire naturelle de la MH en ce qui concerne le moment de l'apparition initiale des symptômes, le diagnostic et la progression est fourni (Figure 2). Cependant, le moment de l'apparition des symptômes, l'ordre de présentation et la trajectoire conséquente des symptômes dans chaque domaine (moteur, cognitif, fonctionnel et comportemental) sont propres à chaque participant. De même, un individu peut être diagnostiqué de différentes manières à différents moments. Enroll-HD collecte donc des données sur une multitude de variables liées au moment des premiers symptômes, à l’apparition de la maladie et au diagnostic (Tableau 1).
Dans Enroll-HD, les dates relatives au début de premiers signes/symptômes sont capturés tels que rapportés selon plusieurs points de vue ; le participant, sa famille et le clinicien/évaluateur Enroll-HD.
Les dates d’apparition relatives à des signes/symptômes spécifiques dans chaque domaine sont également enregistrées. Celles-ci sont complétées du point de vue du clinicien/évaluateur, sur la base de son meilleur jugement. Cela prend en compte les rapports des participants et de leurs familles, les antécédents disponibles à partir des dossiers médicaux, ainsi que le score d'évaluation Enroll-HD.
Le terme 'diagnostic clinique' est utilisé pour désigner l'apparition sans équivoque de symptômes ou de signes attribués à la MH, qui peuvent survenir à des moments très différents pour chaque HDGEC individuel. Dans le protocole Enroll-HD, un jugement basé sur un clinicien quant à l'état de la maladie « manifeste », tel qu'indiqué dans Enroll-HD par catégorie de participants (c.-à-d. hdcat), est basé sur les signes/symptômes n'importe lequel des domaines de la maladie (c.-à-d. moteur, cognitif, comportemental).
Enroll-HD collecte également la date du diagnostic clinique de MH (diagnostic hd). Cette variable représente la date à laquelle un participant est informé par un clinicien que la maladie est évidente. Cependant, cela peut prendre des années après l’apparition réelle des symptômes si le participant n’a pas été vu par un médecin. Si la date du premier diagnostic est inconnue et ne peut être identifiée, diagnostic hd peuvent manquer, même si un clinicien est sûr de son diagnostic de MH symptomatique et a marqué en conséquence hdcat comme manifeste.
Une définition alternative de l’apparition de la maladie, également appelée « manifeste » et largement utilisée dans la littérature sur la MH, concerne la transition d’une MH présymptomatique à une MH symptomatique, basée sur les signes moteur uniquement; c'est ce qu'on appelle début du moteur ou diagnostic moteur. Cette définition est basée sur un score de niveau de confiance diagnostique (DCL) (c.-à-d. diagconf) sur 4, ce qui indique la confiance du clinicien dans le fait que, sur la base de l'évaluation motrice UHDRS, les signes moteurs représentent sans équivoque la MH (confiance ≥ 99%). À condition qu'un participant ne soit pas classé comme hdcat = 'manifeste', ou diagconf = « 4 », à l'entrée dans l'étude (c'est-à-dire lors de la visite de référence), la date de la visite à laquelle l'une ou l'autre de ces variables est mise à jour avec les valeurs ci-dessus peut être utilisée pour indiquer la date d'apparition clinique, comme indiqué respectivement ci-dessus.
Le diagnostic génétique de la MH peut être effectué avant l’apparition des symptômes (connue sous le nom de «test prédictif») ou pour confirmer un diagnostic clinique (appelé «test diagnostique»). Les tests génétiques sont volontaires et les tests sont effectués dans les laboratoires locaux pour certains participants à Enroll-HD, mais pas pour tous. Séparément, tous les participants Enroll-HD subissent un génotypage des répétitions CAG dans un laboratoire de recherche central. Ces résultats sont utilisés uniquement pour recherche par opposition à des fins de diagnostic, et ne sont jamais partagés avec les participants, les investigateurs ou les centres. Comme mentionné précédemment, un individu est un HDGEC s'il a 36 répétitions CAG ou plus. Chez les individus symptomatiques sans antécédents familiaux de MH, le diagnostic clinique est confirmé par des tests génétiques ; par conséquent, la date du test génétique local (c.-à-d. lbdtc) peut être utilisée comme « date du diagnostic clinique » dans de tels cas. Chez les personnes asymptomatiques ayant des antécédents familiaux et qui subissent des tests prédictifs, la date du test génétique peut être utilisée comme « date du diagnostic génétique ». Notez cependant que la date des tests génétiques locaux n'est jamais disponible dans les données Enroll-HD communiquées.
MH Gravité de la maladie
Les étapes importantes dans la progression de la maladie sont quelque peu limitées dans le domaine de la MH. Le Système de classification I-V de Shoulson-Fahn (Shoulson & Fahn, 1979), basé sur le score de capacité fonctionnelle totale (TFC) UHDRS, est traditionnellement utilisé pour classer les individus symptomatiques en stades de la maladie ; I étant le moins sévère, V le plus sévère. Le score TFC est collecté dans Enroll-HD.
La gravité de la maladie peut également être caractérisée en termes de score CAP . Le score CAP signifie CAG-Age-Product (c'est-à-dire le produit de la longueur excessive du CAG et de l'âge) et fournit une mesure de l'exposition cumulée à la HTT mutante (semblable à la mesure des « paquets-années » couramment utilisée dans la recherche sur le tabac comme indicateur (variable) dans l'exposition au tabac). Dans Enroll-HD, le CAP est calculé selon la formule suivante (Warner et al., 2020) :
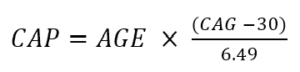
La formule ci-dessus est standardisée de telle sorte que CAP = 100 à l'âge prévu du diagnostic. Pour plus d’informations sur le CAP, consultez «Âge et longueur des CAG dans l'analyse des données MH».
Les évaluations MH dans Enroll-HD
La batterie d'évaluation Enroll-HD a été développée pour permettre une évaluation systématique et complète de la MH. Les éléments de données de base, qui sont obligatoires et doivent être complétés ou revus et mis à jour à chaque visite annuelle, comprennent des informations démographiques sur les participants, les caractéristiques cliniques de la MH, les comorbidités, les traitements liés à la maladie et d'autres thérapies, ainsi que plusieurs évaluations conçues pour évaluer les fonctions motrices, fonctionnelles, les performances comportementales et cognitives, y compris plusieurs éléments de l'Unified Huntington’s Disease Rating Scale (UHDRS®'99). Les évaluations approfondies, facultatives à réaliser à chaque visite, comprennent des tests supplémentaires des fonctions motrices, comportementales et cognitives, ainsi que des évaluations de la qualité de vie et des mesures d'impact sur la santé et l'économie. La page de l'étude Enroll-HD répertorie tous Éléments de données et les évaluations collectés dans Enroll-HD.
Nous discutons ici de deux de ces évaluations – l’UHDRS et la PBA-s – et soulignons quelques bizarreries intéressantes (enfin, notables) dont il faut être conscient en ce qui concerne les données qu’elles génèrent.
UHDRS®'99
L'UHDRS®'99 comprend des éléments moteurs, cognitifs et fonctionnels conçus pour mesurer la progression de la MH dans chaque domaine.
Les signes moteurs sont évalués par le Total Motor Score (TMS). L'échelle comprend 31 éléments, dont l'oculomotricité, la rigidité, la dystonie et la chorée, notés de 0 (aucune anomalie) à 4 (déficience motrice la plus grave). Le score total est la somme des scores de tous les items et varie de 0 à 124, les scores les plus élevés indiquant une déficience motrice accrue. Des précautions doivent être prises lors de l’analyse des données TMS en raison de données très asymétriques cohérentes avec un effet plancher/plafond (Figure 2). Les individus au début de la progression de la MH présenteront moins de signes de déficience motrice et obtiendront un score en haut de l'échelle (c'est-à-dire égal ou proche de zéro). Des méthodes statistiques appropriées, telles que la transformation ou l'utilisation d'une régression binomiale négative, doivent être envisagées.
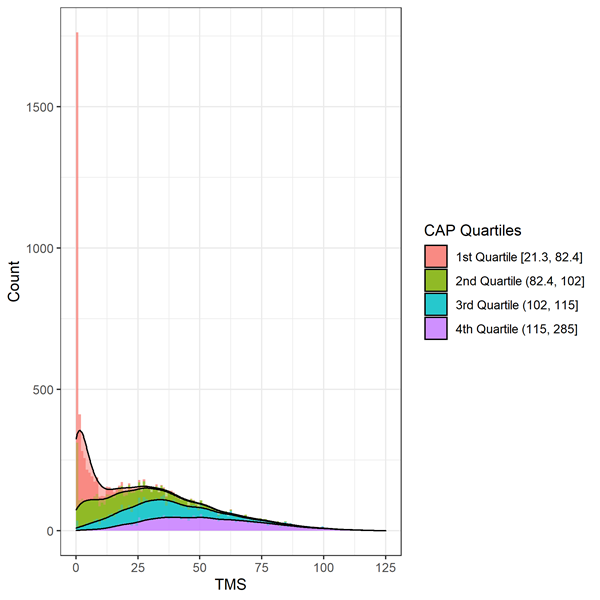
Figure 3. Distribution du TMS de base par Déciles de la PAC en Enroll-HD. Warner et coll. (2020) Formule de score CAP utilisée, qui est standardisée de telle sorte que CAP = 100 à l'âge prévu du diagnostic.
La capacité cognitive est évaluée à l'aide de plusieurs tests qui mesurent des compétences telles que l'attention visuelle et la vitesse de traitement (test de modalités de chiffres symboliques), l'attention de base (tests de lecture de mots et de dénomination des couleurs de Stroop) et l'inhibition de la réponse (test d'interférence de Stroop). Des scores inférieurs à ces tests signifient une diminution des performances cognitives.
Le fonctionnement quotidien est mesuré par trois échelles, la capacité fonctionnelle totale (TFC), l'échelle d'évaluation fonctionnelle (FAS) et l'échelle d'indépendance (IS). Le TFC est la principale évaluation du fonctionnement quotidien dans la MH avec des évaluations dans cinq domaines : la profession, les finances, les tâches domestiques, les activités de la vie quotidienne et le niveau de soins. Le score total du TFC est la somme des scores de chacun de ces domaines et va de 0 (perte de fonction) à 13 (pleine fonction). Comme pour le TFC, des scores plus faibles aux tests FAS (plage = 0 à 25) et IS (plage = 0 à 100) indiquent un déclin de la capacité fonctionnelle. Les mesures fonctionnelles sont également affectées par les effets de plafond avec une inflation aux scores maximum, car les personnes en début de progression de la MH maintiennent souvent leur plein fonctionnement tel qu'évalué par les échelles. Comme pour le TMS, l’utilisation de méthodes statistiques appropriées est encouragée.
Évaluation Problem Behavioral Assessment version courte (PBA-s)
La Problem Behavioral Assessment - version courte (PBA-s) évalue la fréquence et la gravité d'un large éventail de comportements, notamment l'humeur dépressive, l'estime de soi, l'anxiété, les pensées suicidaires, le comportement agressif, l'irritabilité, les persévérations, les comportements compulsifs, les délires, les hallucinations, et l'apathie. Bien que les signes comportementaux soient importants pour les individus et leur qualité de vie, ils ne suivent généralement pas la progression de la MH aussi fortement que les mesures motrices, cognitives et fonctionnelles (Tabrizi et al. 2013 ; Paulsen et al. 2014). Néanmoins, il existe des preuves que l'apathie est associée à la progression de la MH (Kingma et al. 2008 ; McColgan & Tabrizi 2017).