
Introducción a EH y CAG
La EH es una enfermedad neurodegenerativa progresiva, autosómica dominante, causada por una expansión del tracto CAG en el gen HTT ubicado en el cromosoma 4. Los individuos sanos tienen entre 10 y 35 repeticiones CAG, aunque existe cierta evidencia de que aquellos con 27 a 35 repeticiones CAG, conocido como alelos intermedios, pueden mostrar signos de aparición más adelante en la vida (Groen et al. 2010). Portadores de expansión del gen EH (HDGEC) tienen 36 o más repeticiones CAG. Aquellos con 36-39 repeticiones tienen penetrancia reducida, lo que significa que algunos desarrollarán síntomas de la EH a lo largo de su vida, mientras que otros no (McNeil et al. 1997). Las personas con 40 o más repeticiones CAG tienen penetrancia total y desarrollará la EH durante una vida normal.
La edad a la que los HDGEC experimentan la aparición de signos motores se asocia con la duración de las repeticiones CAG, y aquellos con más de 60 repeticiones CAG probablemente presenten signos muy temprano y desarrollen EH juvenil antes de llegar a la edad adulta (Fusilli et al. 2018). En los estudios observacionales de EH, la longitud de CAG que ocurre con más frecuencia es 42, y la mayoría de las HDGEC oscilan entre 40 y 44 CAG. La distribución de longitud CAG para Enroll-HD se muestra en la Figura 1.
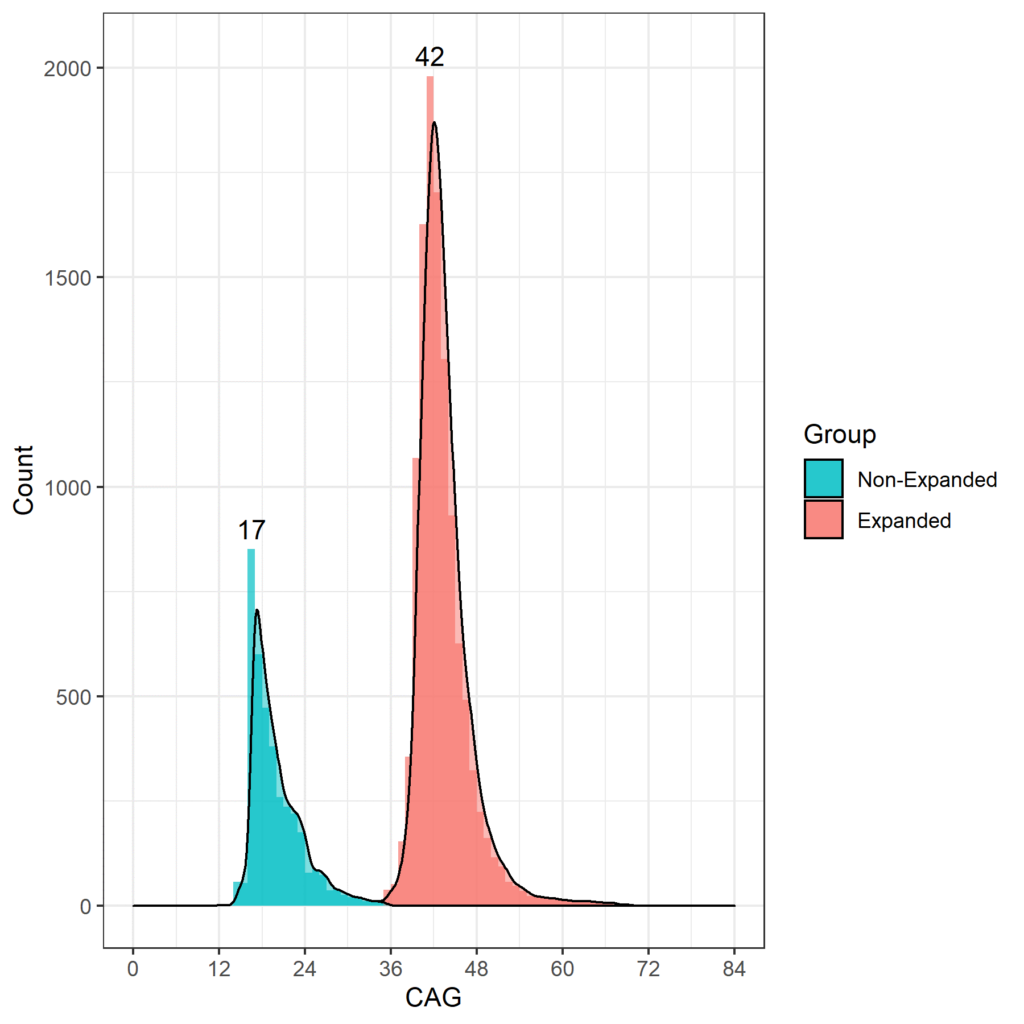
Figura 1. Distribución de longitudes de CAG en la población Enroll-HD. La distribución de las longitudes de repetición CAG en no HDGEC y HDGEC en Enroll-HD se ilustra por separado. Valores modales para cada grupo enumerados explícitamente. (Lanzamiento PDS4; v2018-10-R3).
Inicio y diagnóstico de la EH
El inicio y el diagnóstico de la EH son conceptos importantes de interés crítico para muchos investigadores. En esta sección analizamos estos conceptos complejos en detalle y los matices de las variables que los capturan en el estudio Enroll-HD.
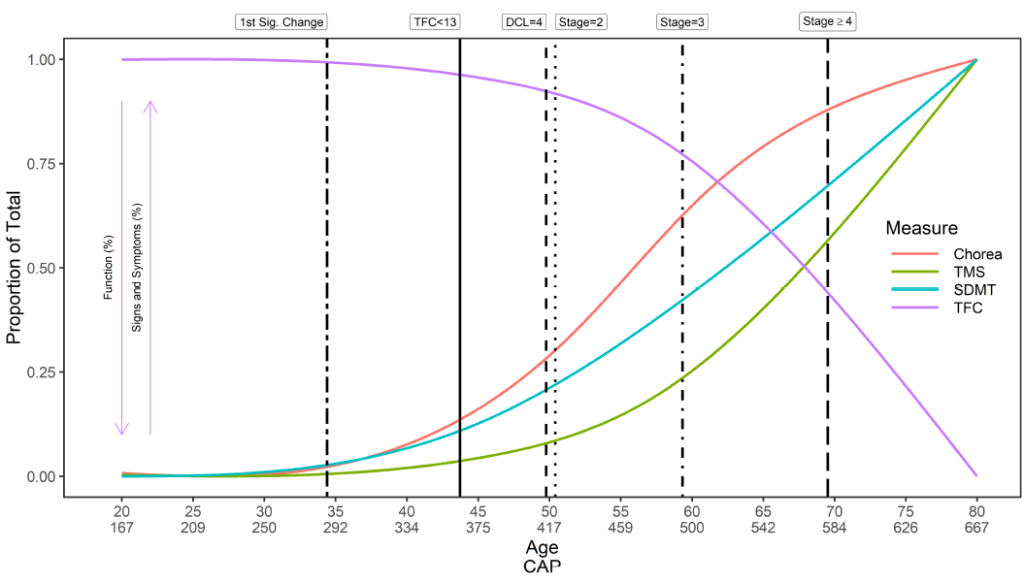
El inicio de la EH es complejo. Se proporciona una descripción general genérica de la historia natural de la EH con respecto al momento de aparición de los síntomas iniciales, el diagnóstico y la progresión (Figura 2). Sin embargo, el momento de aparición de los síntomas, el orden de presentación y la trayectoria consiguiente de los síntomas en cada dominio (motor, cognitivo, funcional y conductual) son únicos para cada participante. De manera similar, un individuo puede ser diagnosticado de diferentes maneras en diferentes momentos. Por lo tanto, Enroll-HD recopila datos sobre una multitud de variables relacionadas con el momento de los síntomas iniciales, la aparición de la enfermedad y el diagnóstico (Tabla 1).
En Enroll-HD, las fechas relacionadas con el inicio de primeros signos/síntomas se capturan tal como se informa desde varias perspectivas; el participante, su familia y el médico/evaluador de Enroll-HD.
También se capturan las fechas de aparición de signos/síntomas específicos en cada dominio. Estos se completan desde la perspectiva del médico/evaluador, según su mejor criterio. Esto tiene en cuenta los informes de los participantes y de la familia, el historial disponible de los registros médicos y la puntuación de la evaluación Enroll-HD.
El término 'diagnostico clinico' se utiliza para denotar la aparición inequívoca de síntomas o signos atribuidos a la EH, que pueden ocurrir en momentos muy diferentes para cada HDGEC individual. En el protocolo Enroll-HD, un juicio clínico sobre el estado de la enfermedad "manifiesta", como se indica en Enroll-HD por categoría de participante (es decir, hdcat), se basa en los signos/síntomas en cualquier de los dominios de la enfermedad (es decir, motor, cognitivo, conductual).
Enroll-HD también captura la fecha del diagnóstico clínico de EH (diagnóstico hd). Esta variable representa la fecha en la que un médico informa a un participante que la enfermedad es evidente. Sin embargo, esto puede ocurrir años después de la aparición real de los síntomas si el participante no ha sido atendido por un médico. Si se desconoce la fecha del primer diagnóstico y no se puede identificar, diagnóstico hd puede faltar, incluso si un médico confía en su diagnóstico de EH sintomática y ha marcado correspondientemente hdcat como manifiesto.
Una definición alternativa de inicio de la enfermedad, también denominada “manifiesta” y ampliamente utilizada en la literatura sobre la EH, se refiere a la transición de la EH presintomática a la sintomática basada en solo señales de motor; esto se conoce como inicio motor o diagnóstico motor. Esta definición se basa en una puntuación del Nivel de confianza diagnóstica (DCL) (es decir, diagconf) de 4, lo que indica la confianza del médico en que, según la evaluación motora UHDRS, los signos motores representan inequívocamente la EH (confianza ≥ 99%). Siempre que un participante no fuera clasificado como hdcat = 'manifiesto', o diagconf = '4', al ingresar al estudio (es decir, en la visita inicial), la fecha de la visita en la que cualquiera de estas variables se actualiza a los valores anteriores se puede utilizar para indicar la fecha de inicio clínico, como se describe respectivamente anteriormente.
Diagnóstico genético La EH se puede realizar antes de la aparición de los síntomas (conocido como “prueba predictiva”) o para confirmar un diagnóstico clínico (conocido como “prueba de diagnóstico”). Las pruebas genéticas son voluntarias y se realizan en laboratorios locales para algunos, pero no para todos, los participantes de Enroll-HD. Por separado, todos los participantes de Enroll-HD se someten a un genotipado repetido CAG en un laboratorio de investigación central. Estos resultados se utilizan únicamente para investigación a diferencia de los fines de diagnóstico, y nunca se comparten con los participantes, investigadores o centros. Como se mencionó anteriormente, un individuo es un HDGEC si tiene 36 o más repeticiones CAG. En individuos sintomáticos sin antecedentes familiares de EH, el diagnóstico clínico se confirma mediante pruebas genéticas; por lo tanto, la fecha de las pruebas genéticas locales (es decir, lbdtc) puede utilizarse como “fecha del diagnóstico clínico” en tales casos. En personas asintomáticas con antecedentes familiares que se someten a pruebas predictivas, la fecha de la prueba genética puede usarse como "fecha del diagnóstico genético". Sin embargo, tenga en cuenta que la fecha de las pruebas genéticas locales nunca está disponible en las publicaciones de datos del Enroll-HD.
Gravedad de la enfermedad de la EH
Los hitos importantes en la progresión de la enfermedad son algo limitados en el campo de la EH. El Sistema de estadificación Shoulson-Fahn IV (Shoulson & Fahn, 1979), basado en la puntuación de Capacidad Funcional Total (TFC) de la UHDRS, se utiliza tradicionalmente para clasificar a los individuos sintomáticos en etapas de la enfermedad; Siendo I el menos grave, V el más grave. La puntuación TFC se captura en Enroll-HD.
La gravedad de la enfermedad también se puede caracterizar en términos de puntuación CAP . La puntuación CAP significa CAG-Edad-Producto (es decir, el producto del exceso de longitud de CAG y edad) y proporciona una medida de la exposición acumulativa a HTT mutante (similar a la métrica de 'paquete-años' comúnmente utilizada en la investigación del tabaco como indicador indirecto). por exposición al tabaco). En Enroll-HD, el CAP se calcula según la siguiente fórmula (Warner et al., 2020):
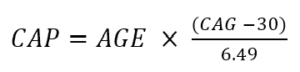
La fórmula anterior está estandarizada de modo que CAP = 100 a la edad esperada de diagnóstico. Para obtener más información sobre CAP, consulte "Edad y longitud CAG en análisis de datos HD”.
Evaluaciones HD en Enroll-HD
La batería de evaluación Enroll-HD fue desarrollada para permitir una evaluación sistemática e integral de la EH. Los componentes de datos básicos, que son obligatorios y deben completarse o revisarse y actualizarse en cada visita anual, incluyen información demográfica de los participantes, características clínicas de la EH, condiciones comórbidas, tratamientos relacionados con enfermedades y otras terapias, y varias evaluaciones diseñadas para evaluar la motricidad, la función, rendimiento conductual y cognitivo, incluidas varias escalas que componen la Escala Unificada de Calificación de la Enfermedad de Huntington (UHDRS®'99). Las evaluaciones extendidas, que son opcionales para completar en cada visita, comprenden pruebas adicionales de función motora, conductual y cognitiva, junto con evaluaciones de la calidad de vida y medidas de impacto económico y de salud. La página de estudio Enroll-HD enumera todos Elementos de datos y evaluaciones capturados en Enroll-HD.
Aquí analizamos dos de estas evaluaciones (la UHDRS y la PBA) y destacamos algunas peculiaridades interesantes (bueno, notables) que se deben tener en cuenta con respecto a los datos que generan.
UHDRS®'99
El UHDRS®'99 comprende componentes motores, cognitivos y funcionales diseñados para medir la progresión de la EH en cada dominio.
Los signos motores se evalúan mediante el Total Motor Score (TMS). La escala se compone de 31 ítems, que incluyen oculomotor, rigidez, distonía y corea, calificados de 0 (sin anomalías) a 4 (deterioro motor más grave). La puntuación total es la suma de las puntuaciones de todos los ítems y oscila entre 0 y 124; las puntuaciones más altas indican un mayor deterioro motor. Se debe tener cuidado al analizar los datos de TMS debido a que los datos están muy sesgados y son consistentes con un efecto suelo/techo (Figura 2). Los individuos en las primeras etapas de la progresión de la EH mostrarán menos signos de deterioro motor y obtendrán una puntuación en la parte superior de la escala (es decir, en o cerca de cero). Deben considerarse métodos estadísticos apropiados, como la transformación o el uso de regresión binomial negativa.
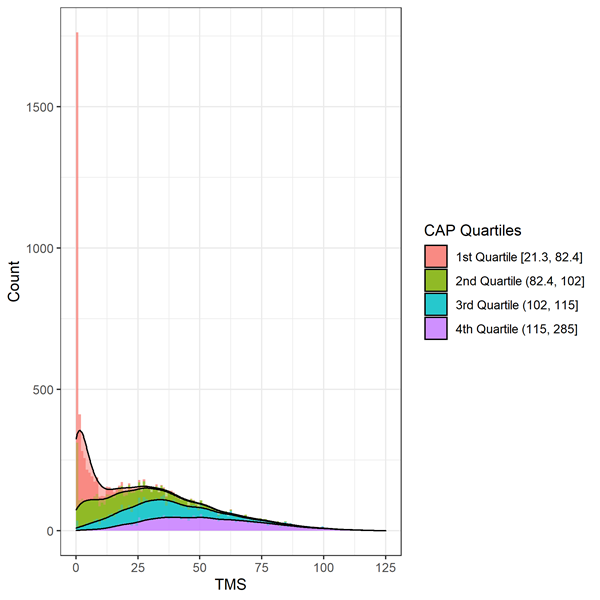
Figura 3. Distribución del TMS basal por Deciles de la PAC en Enroll-HD. Warner et al. (2020) Se utiliza la fórmula de puntuación CAP, que está estandarizada de modo que CAP = 100 en la edad esperada de diagnóstico.
La capacidad cognitiva se evalúa mediante varias pruebas que miden habilidades como la atención visual y la velocidad de procesamiento (Prueba de modalidades de dígitos simbólicos), la atención básica (Pruebas Stroop de lectura de palabras y denominación de colores) y la inhibición de respuesta (Prueba de interferencia Stroop). Las puntuaciones más bajas en estas pruebas significan una disminución del rendimiento cognitivo.
El funcionamiento diario se mide mediante tres escalas, la Capacidad Funcional Total (TFC), la Escala de Evaluación Funcional (FAS) y la Escala de Independencia (IS). La TFC es la evaluación principal del funcionamiento diario en la EH con calificaciones en cinco dominios: ocupación, finanzas, tareas domésticas, actividades de la vida diaria y nivel de atención. La puntuación total de TFC es la suma de las puntuaciones de cada uno de estos dominios y oscila entre 0 (pérdida de función) y 13 (función completa). Al igual que el TFC, las puntuaciones más bajas en el FAS (rango = 0-25) y el IS (rango = 0-100) indican una disminución en la capacidad funcional. Las medidas funcionales también se ven afectadas por efectos de techo con inflación en las puntuaciones máximas porque aquellos que se encuentran en las primeras etapas de la progresión de la EH a menudo mantienen el pleno funcionamiento según lo calificado por las escalas. Al igual que con TMS, se recomienda el uso de métodos estadísticos apropiados.
Evaluación de conducta problemática versión corta
La versión corta de evaluación de conductas problemáticas (PBA-s) evalúa la frecuencia y gravedad de una amplia gama de conductas, incluido el estado de ánimo deprimido, la autoestima, la ansiedad, los pensamientos suicidas, la conducta agresiva, la irritabilidad, la perseveración, las conductas compulsivas, los delirios, las alucinaciones, y apatía. Si bien los signos de comportamiento son importantes para las personas y su calidad de vida, generalmente no se ha encontrado que sigan la progresión de la EH con tanta fuerza como las medidas motoras, cognitivas y funcionales (Tabrizi et al. 2013; Paulsen et al. 2014). Sin embargo, existe evidencia de que la apatía está asociada con la progresión de la EH (Kingma et al. 2008; McColgan & Tabrizi 2017).