
Einführung in HD und CAG
HD ist eine autosomal-dominant vererbte, fortschreitende neurodegenerative Erkrankung, die durch eine Erweiterung des CAG-Trakts im HTT-Gen auf Chromosom 4 verursacht wird. Gesunde Personen haben zwischen 10 und 35 CAG-Wiederholungen, obwohl es Hinweise darauf gibt, dass Personen mit 27 bis 35 CAG-Wiederholungen bekannt als Zwischenallele, kann Anzeichen eines späteren Auftretens im Leben zeigen (Groen et al. 2010). Träger der Huntington-Genexpansion (HDGECs) haben 36 oder mehr CAG-Wiederholungen. Diejenigen mit 36-39 Wiederholungen haben verringerte Penetranz.Das bedeutet, dass einige im Laufe ihres Lebens Huntington-Symptome entwickeln werden, andere jedoch nicht (McNeil et al. 1997). Menschen mit 40 oder mehr CAG-Wiederholungen haben volle Penetranz und werden innerhalb eines normalen Lebens die Huntington-Krankheit entwickeln.
Das Alter, in dem bei HDGECs motorische Anzeichen auftreten, hängt mit der Länge der CAG-Wiederholungen zusammen, und diejenigen mit mehr als 60 CAG-Wiederholungen zeigen wahrscheinlich sehr früh Anzeichen und entwickeln eine juvenile Huntington-Krankheit, bevor sie das Erwachsenenalter erreichen (Fusilli et al. 2018). In Huntington-Beobachtungsstudien beträgt die am häufigsten auftretende CAG-Länge 42, wobei die meisten HDGECs zwischen 40 und 44 CAGs liegen. Die CAG-Längenverteilung für Enroll-HD ist in Abbildung 1 dargestellt.
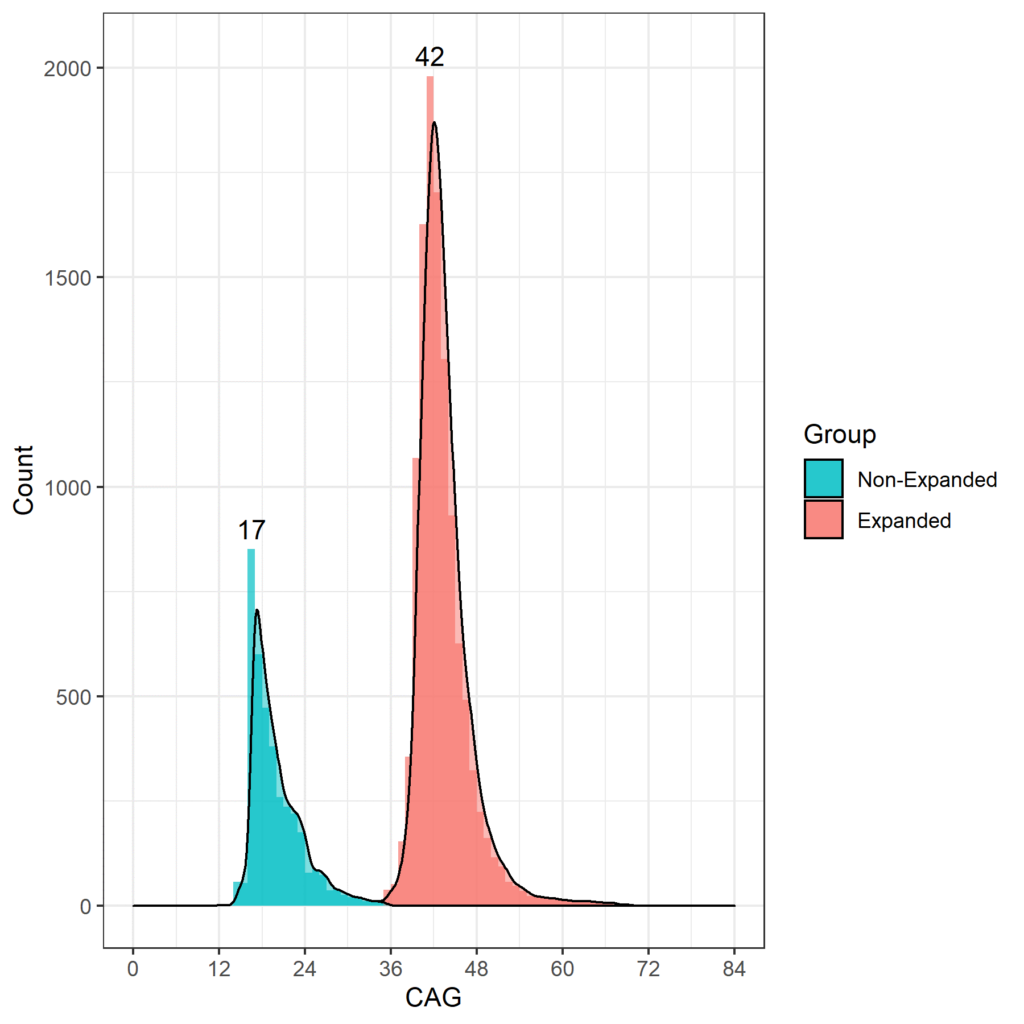
Abbildung 1. Verteilung der CAG-Längen in der Enroll-HD-Population. Verteilung der CAG-Wiederholungslängen in Nicht-HDGECs und HDGECs in Enroll-HD separat dargestellt. Modalwerte für jede Gruppe werden explizit aufgeführt. (PDS4-Version; v2018-10-R3).
Beginn und Diagnose der Huntington-Krankheit
Der Beginn und die Diagnose der Huntington-Krankheit sind wichtige Konzepte, die für viele Forscher von entscheidendem Interesse sind. In diesem Abschnitt besprechen wir diese komplexen Konzepte im Detail und die Nuancen der Variablen, die sie in der Enroll-HD-Studie erfassen.
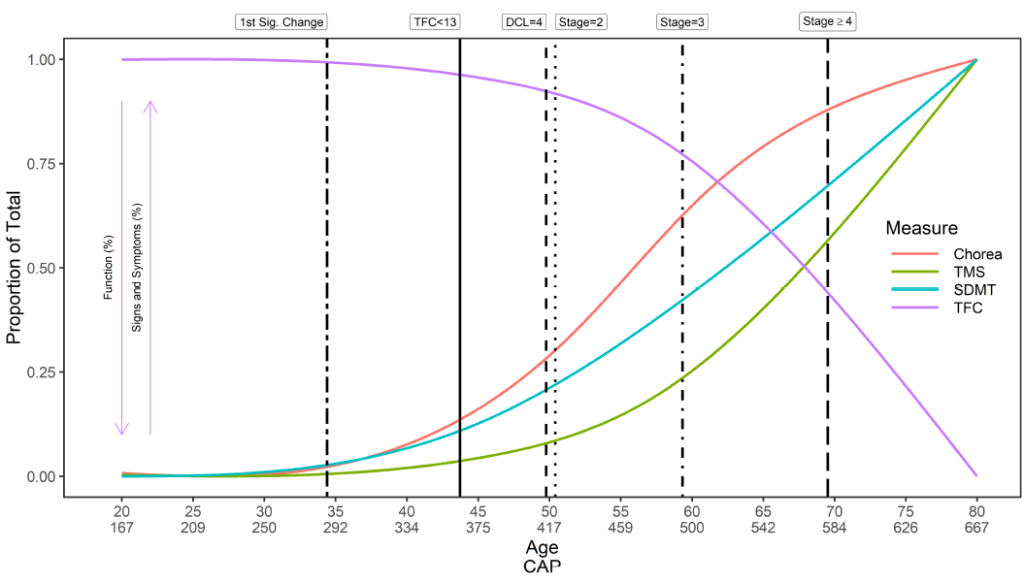
Der Beginn der Huntington-Krankheit ist komplex. Es wird ein allgemeiner Überblick über den natürlichen Verlauf der Huntington-Krankheit im Hinblick auf den Zeitpunkt des ersten Symptombeginns, die Diagnose und das Fortschreiten bereitgestellt (Abbildung 2). Allerdings sind der Zeitpunkt des Symptombeginns, die Reihenfolge des Auftretens und der daraus resultierende Verlauf der Symptome in jedem Bereich – motorisch, kognitiv, funktionell und verhaltensbezogen – für jeden Teilnehmer einzigartig. Ebenso kann eine Person zu unterschiedlichen Zeiten auf unterschiedliche Weise diagnostiziert werden. Enroll-HD sammelt daher Daten zu einer Vielzahl von Variablen im Zusammenhang mit dem Zeitpunkt der ersten Symptome, dem Ausbruch der Krankheit und der Diagnose (Tabelle 1).
In Enroll-HD beziehen sich Daten auf den Beginn von erste Anzeichen/Symptome werden wie berichtet aus mehreren Perspektiven erfasst; der Teilnehmer, seine Familie und der Enroll-HD-Kliniker/Bewerter.
Außerdem werden die Eintrittsdaten für bestimmte Anzeichen/Symptome in den einzelnen Bereichen erfasst. Diese werden aus der Sicht des Klinikers/Bewerters nach bestem Ermessen durchgeführt. Dabei werden Teilnehmer- und Familienberichte, verfügbare Krankengeschichten aus Krankenakten sowie das Enroll-HD-Bewertungsergebnis berücksichtigt.
Der Begriff 'klinische Diagnose' wird verwendet, um das eindeutige Auftreten von Symptomen oder Anzeichen zu bezeichnen, die der Huntington-Krankheit zugeschrieben werden und bei jedem einzelnen HDGEC zu sehr unterschiedlichen Zeiten auftreten können. Im Enroll-HD-Protokoll erfolgt eine ärztliche Beurteilung des „manifesten“ Krankheitsstatus, wie in Enroll-HD nach Teilnehmerkategorie angegeben (d. h. hdcat), basierend auf Anzeichen/Symptomen in einer der Krankheitsdomänen (z. B. motorisch, kognitiv, verhaltensbezogen).
Enroll-HD erfasst auch das Datum der klinischen Huntington-Diagnose (hddiagn). Diese Variable stellt das Datum dar, an dem ein Teilnehmer von einem Arzt darüber informiert wird, dass die Krankheit offensichtlich ist. Dies kann jedoch Jahre nach dem tatsächlichen Auftreten der Symptome erfolgen, wenn der Teilnehmer nicht von einem Arzt untersucht wurde. Wenn das Datum der Erstdiagnose unbekannt ist und nicht identifiziert werden kann, hddiagn können fehlen, selbst wenn ein Arzt von seiner Diagnose einer symptomatischen Huntington-Krankheit überzeugt ist und entsprechende Markierungen vorgenommen hat hdcat als manifest.
Eine alternative Definition des Krankheitsausbruchs, die auch als „manifest“ bezeichnet wird und in der Huntington-Literatur weit verbreitet ist, betrifft den Übergang von der präsymptomatischen zur symptomatischen Huntington-Krankheit, basierend nur auf Motorsymptomen; das nennt man motorischer Beginn oder motorische Diagnose. Diese Definition basiert auf einem DCL-Score (Diagnostic Confidence Level) (d. h. diagconf) von 4, was auf die Zuversicht eines Arztes hinweist, dass die motorischen Zeichen basierend auf der UHDRS-Motorbewertung eindeutig die Huntington-Krankheit darstellen (≥ 99%-Konfidenz). Vorausgesetzt, ein Teilnehmer wurde nicht als eingestuft hdcat = 'manifestieren', oder diagconf = „4“, bei Studieneintritt (d. h. beim Basisbesuch) kann das Datum des Besuchs, bei dem eine dieser Variablen auf die oben genannten Werte aktualisiert wird, zur Angabe des Datums des klinischen Beginns verwendet werden, wie jeweils oben beschrieben.
Genetische Diagnose der Huntington-Krankheit kann vor dem Einsetzen der Symptome durchgeführt werden (bekannt als „Vorhersagetest“) oder zur Bestätigung einer klinischen Diagnose (bekannt als „Diagnosetest“). Gentests sind freiwillig und werden für einige, aber nicht alle Enroll-HD-Teilnehmer in örtlichen Labors durchgeführt. Alle Enroll-HD-Teilnehmer werden separat einer CAG-Wiederholungsgenotypisierung in einem zentralen Forschungslabor unterzogen. Diese Ergebnisse werden ausschließlich für verwendet Forschung im Gegensatz zu diagnostischen Zwecken und werden niemals an Teilnehmer, Ermittler oder Standorte weitergegeben. Wie bereits erwähnt, ist eine Person ein HDGEC, wenn sie 36 oder mehr CAG-Wiederholungen aufweist. Bei symptomatischen Personen ohne Familiengeschichte von Huntington-Krankheit, die klinische Diagnose wird durch Gentests bestätigt; daher das Datum des lokalen Gentests (d. h. lbdtc) kann in solchen Fällen als „Datum der klinischen Diagnose“ verwendet werden. Bei asymptomatischen Personen mit Familienanamnese, die sich einem prädiktiven Test unterziehen, kann das Datum des Gentests als „Datum der genetischen Diagnose“ verwendet werden. Beachten Sie jedoch, dass das Datum lokaler Gentests in Enroll-HD-Datenveröffentlichungen niemals verfügbar gemacht wird.
Schweregrad der HD-Erkrankung
Wichtige Meilensteine im Krankheitsverlauf sind im Bereich der Huntington-Krankheit etwas begrenzt. Das Shoulson-Fahn IV-Stufensystem (Shoulson & Fahn, 1979), basierend auf dem UHDRS Total Functional Capacity (TFC) Score, wird traditionell verwendet, um symptomatische Personen in Krankheitsstadien einzuteilen; I ist am wenigsten schwerwiegend, V am schwerwiegendsten. Der TFC-Score wird in Enroll-HD erfasst.
Der Schweregrad der Erkrankung kann auch durch den CAP-Score charakterisiert werden. Der CAP-Score steht für CAG-Age-Product (d. h. das Produkt aus überschüssiger CAG-Länge und Alter) und ist ein Maß für die kumulative Exposition gegenüber mutiertem HTT (ähnlich wie die in der Tabakforschung übliche Metrik der "Packungsjahre" als Stellvertreter für die Tabakexposition). In Enroll-HD wird CAP nach der folgenden Formel berechnet (Warner et al., 2020):
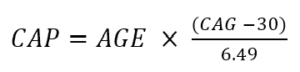
Die obige Formel ist so standardisiert, dass CAP = 100 im erwarteten Diagnosealter ist. Weitere Informationen zu CAP finden Sie unter „Alter und CAG-Länge in der HD-Datenanalyse“.
HD-Bewertungen in Enroll-HD
Die Enroll-HD-Bewertungsbatterie wurde entwickelt, um eine systematische und umfassende Bewertung der Huntington-Krankheit zu ermöglichen. Zu den Kerndatenkomponenten – die obligatorisch sind und bei jedem jährlichen Besuch vervollständigt oder überprüft und aktualisiert werden müssen – gehören demografische Informationen der Teilnehmer, klinische Merkmale der Huntington-Krankheit, komorbide Zustände, krankheitsbedingte Behandlungen und andere Therapien sowie verschiedene Beurteilungen zur Beurteilung von Motorik, Funktion, Verhaltens- und kognitive Leistung, einschließlich mehrerer Komponentenskalen der Unified Huntington's Disease Rating Scale (UHDRS®'99). Erweiterte Untersuchungen – die bei jedem Besuch optional durchgeführt werden können – umfassen zusätzliche Tests der motorischen, Verhaltens- und kognitiven Funktion sowie Beurteilungen der Lebensqualität und Maßnahmen zur Messung der gesundheitlichen und wirtschaftlichen Auswirkungen. Die Enroll-HD-Studienseite listet alle auf Datenelemente und Bewertungen erfasst in Enroll-HD.
Hier besprechen wir zwei dieser Bewertungen – UHDRS und PBA-s – und heben einige spannende (naja, bemerkenswerte) Eigenheiten hervor, die man im Hinblick auf die von ihnen generierten Daten beachten sollte.
UHDRS®'99
Das UHDRS®'99 umfasst motorische, kognitive und funktionelle Komponenten, die darauf ausgelegt sind, das Fortschreiten der Huntington-Krankheit in jedem Bereich zu messen.
Motorische Zeichen werden anhand des Total Motor Score (TMS) bewertet. Die Skala besteht aus 31 Items, darunter Okulomotorik, Rigidität, Dystonie und Chorea, mit einer Bewertung von 0 (keine Anomalien) bis 4 (schwerste motorische Beeinträchtigung). Der Gesamtscore ist die Summe der Scores für alle Items und liegt zwischen 0 und 124, wobei höhere Scores auf eine stärkere motorische Beeinträchtigung hinweisen. Bei der Analyse von TMS-Daten ist Vorsicht geboten, da die Daten stark verzerrt sind und auf einen Boden-/Deckeneffekt hinweisen (Abbildung 2). Personen zu Beginn der Huntington-Progression zeigen weniger Anzeichen einer motorischen Beeinträchtigung und erzielen einen Wert am oberen Ende der Skala (dh bei oder nahe Null). Geeignete statistische Methoden, wie etwa die Transformation oder die Verwendung einer negativen Binomialregression, sollten in Betracht gezogen werden.
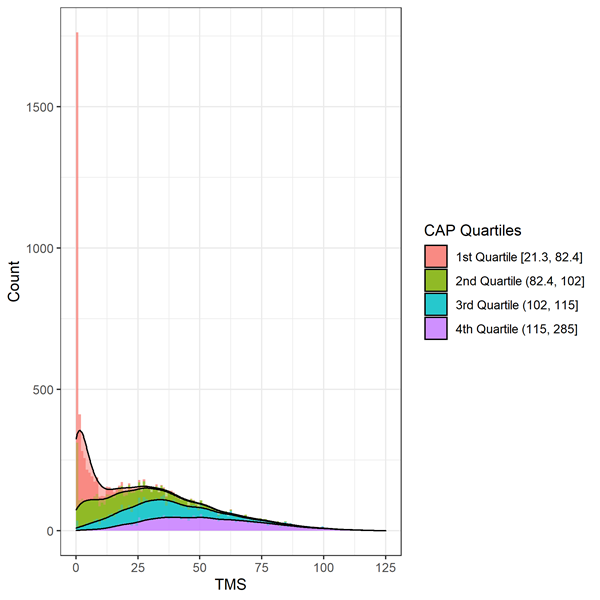
Figur 3. Verteilung des Basis-TMS nach CAP-Dezile im Enroll-HD. Warner et al. (2020) Verwendete CAP-Score-Formel, die so standardisiert ist, dass CAP = 100 im erwarteten Diagnosealter.
Die kognitiven Fähigkeiten werden anhand mehrerer Tests beurteilt, die Fähigkeiten wie visuelle Aufmerksamkeit und Verarbeitungsgeschwindigkeit (Symbol-Ziffer-Modalitätstest), grundlegende Aufmerksamkeit (Stroop-Wortlese- und Farbbenennungstests) und Reaktionshemmung (Stroop-Interferenztest) messen. Niedrigere Ergebnisse bei diesen Tests bedeuten eine verminderte kognitive Leistung.
Die tägliche Leistungsfähigkeit wird anhand von drei Skalen gemessen: der Total Functional Capacity (TFC), der Functional Assessment Scale (FAS) und der Independence Scale (IS). Der TFC ist die primäre Beurteilung der täglichen Funktionsfähigkeit bei der Huntington-Krankheit mit Bewertungen in fünf Bereichen: Beruf, Finanzen, Hausarbeiten, Aktivitäten des täglichen Lebens und Pflegegrad. Der TFC-Gesamtscore ist die Summe der Scores aus jedem dieser Bereiche und reicht von 0 (Funktionsverlust) bis 13 (vollständige Funktion). Wie beim TFC deuten niedrigere Werte beim FAS (Bereich = 0–25) und IS (Bereich = 0–100) auf eine Verschlechterung der Funktionsfähigkeit hin. Die Funktionsmessungen werden auch durch Obergrenzeneffekte beeinflusst, wenn die Inflation bei den maximalen Werten liegt, da diejenigen, die sich zu Beginn der Huntington-Krankheitsprogression befinden, häufig ihre volle Funktionsfähigkeit gemäß den Skalen beibehalten. Wie bei TMS wird die Verwendung geeigneter statistischer Methoden empfohlen.
Kurzfassung zur Bewertung des Problemverhaltens
Die Problem Behavioral Assessment Short Version (PBA-s) bewertet die Häufigkeit und Schwere einer Vielzahl von Verhaltensweisen, darunter depressive Verstimmung, Selbstwertgefühl, Angstzustände, Selbstmordgedanken, aggressives Verhalten, Reizbarkeit, Beharrlichkeit, zwanghaftes Verhalten, Wahnvorstellungen, Halluzinationen, und Apathie. Obwohl die Verhaltensmerkmale für den Einzelnen und seine Lebensqualität wichtig sind, wurde im Allgemeinen nicht festgestellt, dass sie das Fortschreiten der Huntington-Krankheit so stark beeinflussen wie die motorischen, kognitiven und funktionellen Maßnahmen (Tabrizi et al. 2013; Paulsen et al. 2014). Dennoch gibt es Hinweise darauf, dass Apathie mit dem Fortschreiten der Huntington-Krankheit verbunden ist (Kingma et al. 2008; McColgan & Tabrizi 2017).