
Introduzione all'HD e al CAG
La MH è una malattia neurodegenerativa progressiva autosomica dominante causata da un'espansione del tratto CAG nel gene HTT situato sul cromosoma 4. Gli individui sani hanno tra 10 e 35 ripetizioni CAG, sebbene vi siano prove che quelli con 27-35 ripetizioni CAG, conosciuto come alleli intermedi, possono mostrare segni di esordio più tardi nella vita (Groen et al. 2010). Portatori di espansione del gene HD (HDGEC) hanno 36 o più ripetizioni CAG. Quelli con 36-39 ripetizioni lo hanno fatto penetranza ridotta, il che significa che alcuni svilupperanno i sintomi della MH nel corso della loro vita, mentre altri no (McNeil et al. 1997). Le persone con 40 o più ripetizioni CAG hanno penetranza completa e svilupperanno la MH nel corso della loro vita.
L'età in cui gli HDGEC manifestano l'esordio dei segni motori è associata alla lunghezza delle ripetizioni CAG e quelli con più di 60 ripetizioni CAG hanno probabilità di mostrare segni molto presto e sviluppare MH giovanile prima di raggiungere l'età adulta (Fusilli et al. 2018). Negli studi osservazionali sulla MH la lunghezza CAG più frequente è 42, con la maggior parte degli HDGEC che vanno da 40 a 44 CAG. La distribuzione della lunghezza CAG per Enroll-HD è mostrata nella Figura 1.
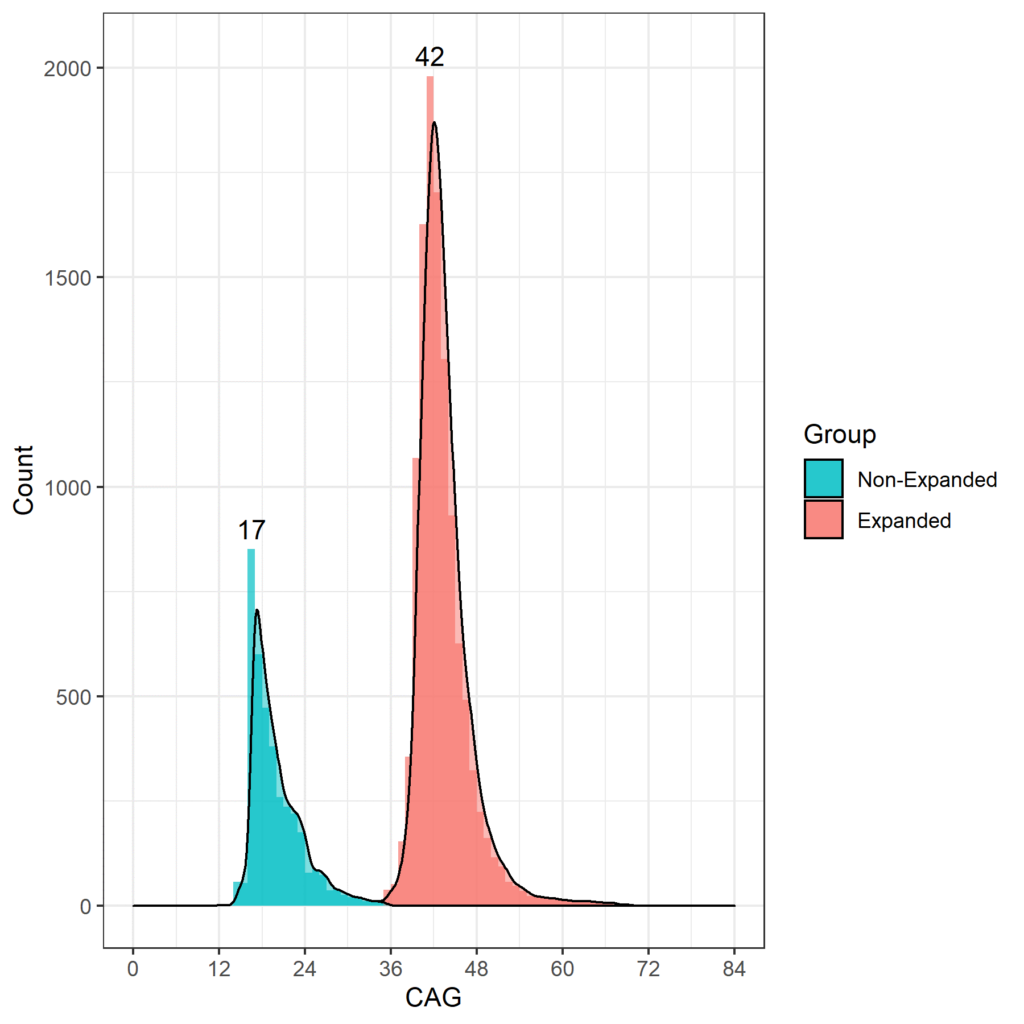
Figura 1. Distribuzione delle lunghezze CAG nella popolazione Enroll-HD. Distribuzione delle lunghezze di ripetizione CAG in non HDGEC e HDGEC in Enroll-HD illustrata separatamente. Valori modali per ciascun gruppo elencati esplicitamente. (Versione PDS4; v2018-10-R3).
Insorgenza e diagnosi della MH
L'esordio e la diagnosi della MH sono concetti importanti di interesse critico per molti ricercatori. In questa sezione discutiamo in dettaglio questi concetti complessi e le sfumature delle variabili che sono analizzate nello studio Enroll-HD.
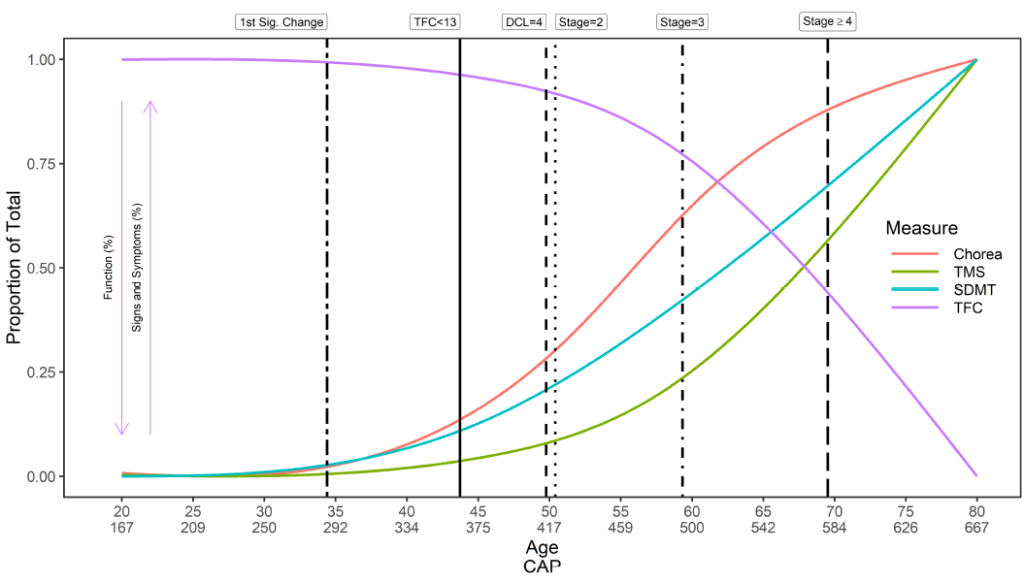
L'esordio della MH è complesso. Viene fornita una panoramica generica della storia naturale della MH per quanto riguarda i tempi di insorgenza dei sintomi iniziali, diagnosi e progressione (Figura 2). Tuttavia, i tempi di insorgenza dei sintomi, l’ordine di presentazione e la conseguente traiettoria dei sintomi in ciascun dominio – motorio, cognitivo, funzionale e comportamentale – sono unici per ciascun partecipante. Allo stesso modo, un individuo può essere diagnosticato in modi diversi in momenti diversi. Enroll-HD raccoglie quindi dati su una moltitudine di variabili relative alla tempistica dei sintomi iniziali, all'insorgenza della malattia e alla diagnosi (Tabella 1).
In Enroll-HD, le date relative all'inizio del primi segnali/sintomi vengono catturati come riportati da diverse prospettive; il partecipante, la sua famiglia e il clinico/valutatore Enroll-HD.
Vengono inoltre acquisite le date di insorgenza relative a segni/sintomi specifici in ciascun dominio. Questi vengono completati dal punto di vista del medico/valutatore, in base al loro miglior giudizio. Ciò tiene conto dei resoconti dei partecipanti e dei familiari, della storia disponibile dalle cartelle cliniche e del punteggio di valutazione Enroll-HD.
Il termine 'diagnosi clinica' è usato per denotare l'insorgenza inequivocabile di sintomi o segni attribuiti alla MH, che possono verificarsi in tempi molto diversi per ogni singolo HDGEC. Nel protocollo Enroll-HD un giudizio basato sul medico sullo stato della malattia "manifesto", come indicato in Enroll-HD per categoria di partecipante (ovvero, hdcat), si basa sui segni/sintomi presenti in Qualunque dei domini della malattia (cioè motorio, cognitivo, comportamentale).
Enroll-HD comprende anche i dati della diagnosi clinica di MH (hddiag). Questa variabile rappresenta la data in cui un partecipante viene informato da un medico che la malattia è evidente. Tuttavia, ciò può avvenire anni dopo l’effettiva comparsa dei sintomi se il partecipante non è stato visto da un medico. Se la data della prima diagnosi è sconosciuta e non può essere identificata, hddiag può mancare, anche se un medico è fiducioso nella diagnosi di HD sintomatica e l'ha contrassegnata di conseguenza hdcat come manifesto.
Una definizione alternativa di esordio della malattia, definita anche “manifesta” e ampiamente utilizzata nella letteratura sulla MH, riguarda la transizione dalla MH presintomatica a quella sintomatica basata sulla solo segni motori; questo è noto come esordio motorio O diagnosi motoria. Questa definizione si basa sul punteggio del livello di fiducia diagnostica (DCL) (vale a dire, diagconf) di 4, che indica la confidenza del medico che, sulla base della valutazione motoria UHDRS, i segni motori rappresentano inequivocabilmente la MH (confidenza ≥ 99%). A condizione che un partecipante non sia stato classificato come hdcat = 'manifesto', o diagconf = '4', all'ingresso nello studio (cioè alla visita basale), la data della visita in cui una di queste variabili viene aggiornata ai valori sopra indicati può essere utilizzata per indicare la data di esordio clinico, come delineato rispettivamente sopra.
Diagnosi genetica della MH può essere effettuata prima della comparsa dei sintomi (noto come “test predittivo") o per confermare una diagnosi clinica (nota come "Test diagnostico"). I test genetici sono volontari e i test vengono completati presso i laboratori locali per alcuni, ma non tutti, i partecipanti Enroll-HD. Separatamente, tutti i partecipanti allo studio Enroll-HD vengono sottoposti alla misurazione del numero di CAG presso un laboratorio di ricerca centrale. Questi risultati vengono utilizzati esclusivamente per ricerca al contrario degli scopi diagnostici e non vengono mai condivisi con partecipanti, ricercatori o siti. Come accennato in precedenza, un individuo è un HDGEC se ha 36 o più ripetizioni CAG. Negli individui sintomatici senza storia familiare di MH, la diagnosi clinica è confermata dai test genetici; quindi, data del test genetico locale (cioè, lbdtc) può essere utilizzato come “data della diagnosi clinica” in questi casi. Negli individui asintomatici con storia familiare sottoposti a test predittivi, la data del test genetico può essere utilizzata come “data della diagnosi genetica”. Si noti tuttavia che la data dei test genetici locali non è mai resa disponibile nei rilasci di dati Enroll-HD.
Gravità della malattia HD
I traguardi importanti nella progressione della malattia sono in qualche modo limitati nel campo della MH. IL Sistema di stadiazione Shoulson-Fahn IV (Shoulson & Fahn, 1979), basato sul punteggio UHDRS della capacità funzionale totale (TFC), è tradizionalmente utilizzato per classificare gli individui sintomatici in stadi della malattia; il punteggio I significa meno grave, V è il più grave. Il punteggio TFC viene misurato in Enroll-HD.
La gravità della malattia può anche essere caratterizzata in termini di punteggio CAP . Il punteggio CAP sta per CAG-Age-Product (ovvero il prodotto tra lunghezza ed età CAG in eccesso) e fornisce una misura dell'esposizione cumulativa all'HTT mutante (simile alla metrica "pacchetto-anno" comunemente utilizzata nella ricerca sul tabacco come proxy per esposizione al tabacco). In Enroll-HD, la CAP viene calcolata secondo la seguente formula (Warner et al., 2020):
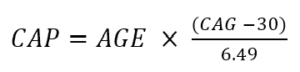
La formula di cui sopra è standardizzata in modo tale che CAP = 100 all'età prevista per la diagnosi. Per maggiori informazioni sulla CAP, consulta “Età e lunghezza CAG nell'analisi dei dati HD”.
Valutazioni HD in Enroll-HD
La batteria di valutazione Enroll-HD è stata sviluppata per consentire una valutazione sistematica e completa della MH. I componenti principali dei dati, che sono obbligatori e devono essere completati o rivisti e aggiornati ad ogni visita annuale, includono informazioni demografiche sui partecipanti, caratteristiche cliniche della MH, condizioni di comorbilità, trattamenti correlati alla malattia e altre terapie, e diverse valutazioni progettate per valutare le capacità motorie, funzionali, prestazioni comportamentali e cognitive, comprese diverse scale componenti della Unified Huntington's Disease Rating Scale (UHDRS®'99). Le valutazioni estese, che possono essere completate facoltativamente a ogni visita, comprendono test aggiuntivi delle funzioni motorie, comportamentali e cognitive, insieme a valutazioni della qualità della vita e misure di impatto economico e sanitario. La pagina dello studio Enroll-HD li elenca tutti Elementi di dati e valutazioni raccolti in Enroll-HD.
Qui discutiamo due di queste valutazioni, UHDRS e PBA, ed evidenziamo alcune interessanti (beh, notevoli) peculiarità di cui essere consapevoli per quanto riguarda i dati che generano.
UHDRS®'99
L'UHDRS®'99 comprende componenti motori, cognitivi e funzionali progettati per misurare la progressione della MH in ciascun dominio.
I segni motori vengono valutati mediante il punteggio motorio totale (TMS). La scala comprende 31 item, tra cui oculomotore, rigidità, distonia e corea, classificati da 0 (nessuna anomalia) a 4 (danno motorio più grave). Il punteggio totale è la somma dei punteggi di tutti gli item e varia da 0 a 124, con punteggi più alti che indicano un aumento della compromissione motoria. È necessario prestare attenzione quando si analizzano i dati TMS a causa di dati altamente distorti coerenti con un effetto pavimento/soffitto (Figura 2). Gli individui all'inizio del corso della progressione della MH mostreranno meno segni di compromissione motoria e avranno un punteggio in cima alla scala (cioè pari o vicino allo zero). Dovrebbero essere presi in considerazione metodi statistici appropriati, come la trasformazione o l’uso della regressione binomiale negativa.
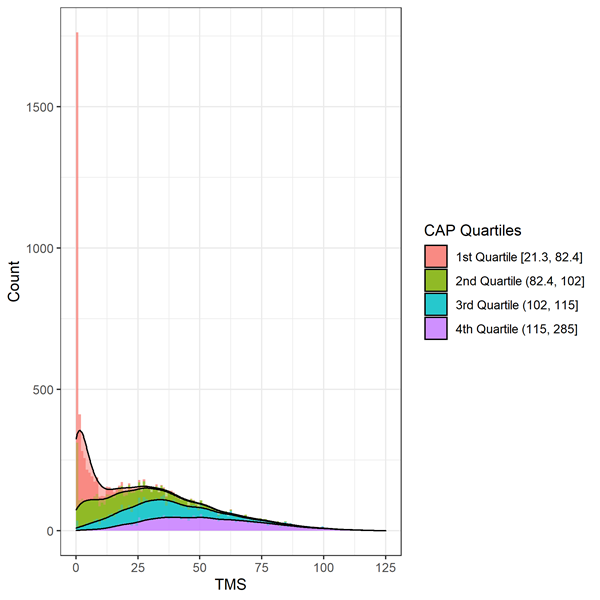
Figura 3. Distribuzione della TMS al basale per decili della PAC nell'Enroll-HD. Warner et al. (2020) È stata utilizzata la formula del punteggio CAP, standardizzata in modo tale che CAP = 100 all'età prevista per la diagnosi.
L'abilità cognitiva viene valutata utilizzando diversi test che misurano capacità come l'attenzione visiva e la velocità di elaborazione (Symbol Digit Modalities Test), l'attenzione di base (Stroop Word Reading and Color Naming Tests) e l'inibizione della risposta (Stroop Interference Test). Punteggi più bassi in questi test indicano una diminuzione delle prestazioni cognitive.
Il funzionamento quotidiano è misurato da tre scale, la capacità funzionale totale (TFC), la scala di valutazione funzionale (FAS) e la scala di indipendenza (IS). Il TFC è la valutazione primaria del funzionamento quotidiano nella MH con valutazioni in cinque ambiti: occupazione, finanze, faccende domestiche, attività della vita quotidiana e livello di assistenza. Il punteggio totale del TFC è la somma dei punteggi di ciascuno di questi domini e varia da 0 (perdita di funzione) a 13 (piena funzione). Come il TFC, i punteggi più bassi su FAS (intervallo = 0–25) e IS (intervallo = 0–100) indicano un declino dell'abilità funzionale. Anche le misure funzionali sono influenzate anche dagli effetti pavimento/soffitto perché i pazienti all'inizio della MH spesso mantengono il pieno funzionamento come valutato dalle scale. Come nel caso del TMS, è incoraggiato l’uso di metodi statistici appropriati.
Versione breve sulla valutazione comportamentale del problema
La Problem Behavioral Assessment Short Version (PBA-s) valuta la frequenza e la gravità di una vasta gamma di comportamenti, tra cui umore depresso, autostima, ansia, pensieri suicidari, comportamento aggressivo, irritabilità, perseverazione, comportamenti compulsivi, deliri, allucinazioni, e apatia. Sebbene i segni comportamentali siano importanti per gli individui e per la loro qualità di vita, in genere non è stato riscontrato che siano in linea con la progressione della MH in modo così forte come le misure motorie, cognitive e funzionali (Tabrizi et al. 2013; Paulsen et al. 2014). Tuttavia, esistono prove che l’apatia è associata alla progressione della MH (Kingma et al. 2008; McColgan & Tabrizi 2017).